
Where exactly do new infections with the coronavirus come from? To answer that question, geneticists look at the pedigree of the virus. Marion Koopmans explains. (in Dutch)
Uit NRC, 13 maart 2020. Sander Voormolen
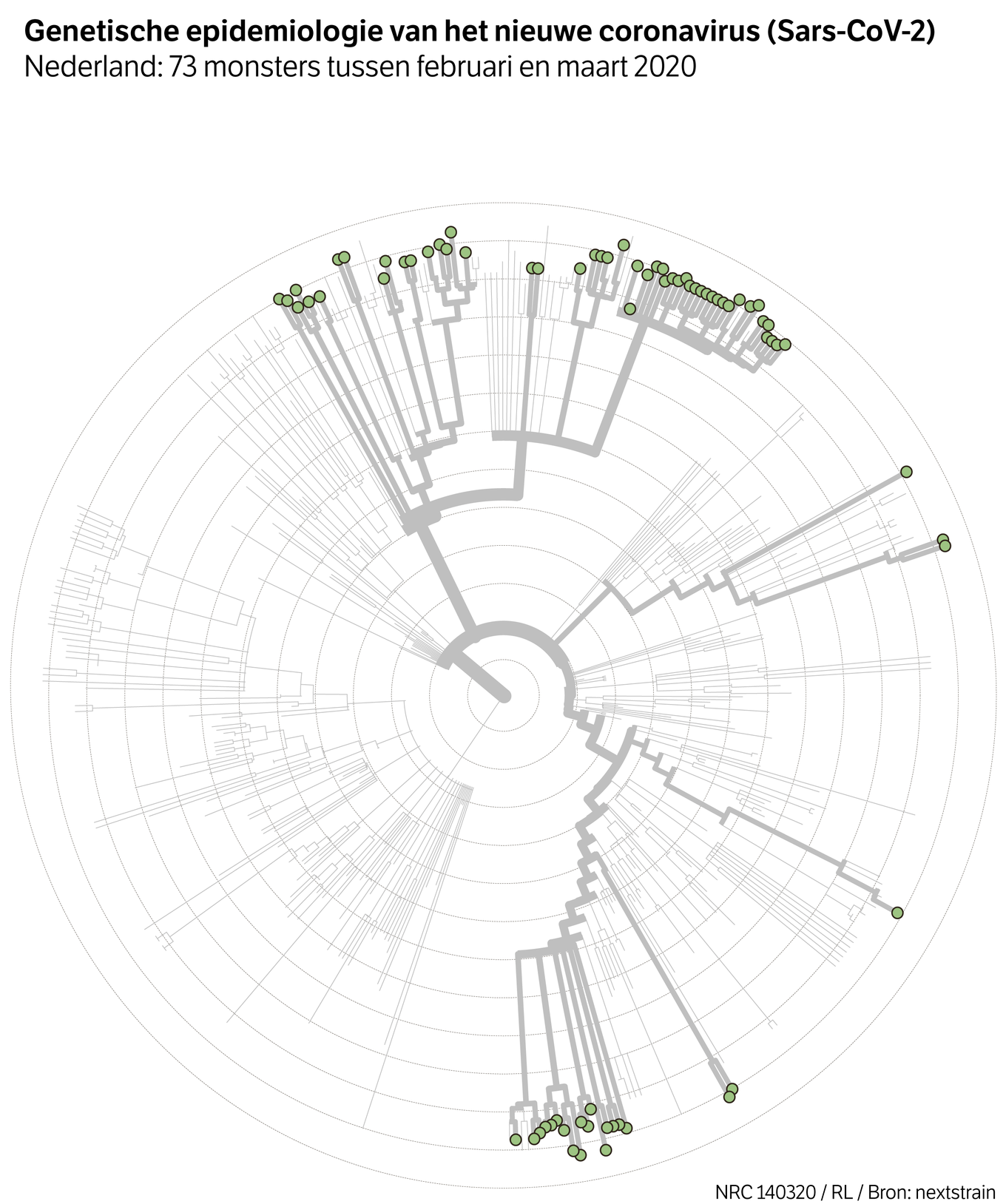
Het nieuwe coronavirus dat nu dik twee weken in Nederland is, is verschillende keren in ons land geïntroduceerd. Dat is te zien aan de stamboom van het coronavirus die genetici hebben samengesteld. Genetici maken deze stamboomanalyses tegenwoordig razendsnel, en kunnen zo het verloop van de epidemie bijna live volgen.
De uitbraak van het nieuwe coronavirus wordt onderzocht door mensen met klachten te testen op de aanwezigheid van het virus. De test detecteert de aanwezigheid van een klein kenmerkend stukje virus-rna. Terwijl de GGD’s hun best doen om contacten van mensen die positief testen te traceren, speuren genetici naar de herkomst van de besmettingen door het complete erfelijke materiaal van het virus te analyseren. Het geeft een belangrijke extra aanwijzing over hoe de epidemie zich ontvouwt. Het kan bijvoorbeeld antwoord geven op de vraag of de uitbraak nog steeds gevoed wordt vanuit buitenlandse brongebieden, of dat het virus zich al zo heeft geworteld in de bevolking dat er een nieuwe, eigen virushaard is ontstaan. Die informatie speelde ook een rol bij de afwegingen voor de nieuwe coronamaatregelen die de Nederlandse overheid deze week nam.
Viroloog Marion Koopmans van het Erasmus MC in Rotterdam en haar team hebben inmiddels al tientallen genetische vingerafdrukken van Nederlandse coronavirussen verzameld. Kleine wijzigingen in de erfelijke code van het virus gebruiken zij om een stamboom te maken van het virus. Via deze analyse konden de onderzoekers zien dat veel van de infecties met het nieuwe coronavirus in Nederland inderdaad veel overeenkomsten hebben met virusvarianten die circuleren in Noord-Italië. Een kleiner deel overlapt met virussen uit brongebieden in Duitsland, Frankrijk en Zuid-Korea.
„Op deze manier willen we de bronopsporing helpen”, zegt Koopmans. Dat gaat vooral via contactonderzoek van mensen met een bevestigde besmetting, door mensen systematisch uit te vragen waar zij geweest zijn en met wie zij in de laatste veertien dagen (intensief) contact hebben gehad. Maar lang niet altijd lukt het zo te traceren waar iemand het virus heeft opgelopen. „De genetische data die wij hebben verzameld hebben we gekoppeld aan de informatie die wij kregen van GGD en RIVM. Dat levert soms nieuwe aanwijzingen op over waar men de bron kan zoeken”, zegt Koopmans. „Maar deze analysemethode om patronen van een epidemie te monitoren is zeker niet fool proof. Het is een experimentele methode, je kunt er niet alles uit afleiden en je moet oppassen voor grote conclusies.”
Begin deze week voegde het team uit Rotterdam de eerste 25 sequenties van het virus toe aan de internationale databank die gebruikt wordt door Nextstrain. Donderdag kwamen er weer 48 bij „We hebben er nog meer, die voegen we later toe”, zegt Koopmans. In Nextstrain krijgen de Nederlandse virussen tussen buitenlandse varianten een duidelijke plek. De Nederlandse sequenties zitten in de stamboom dicht bij elkaar, in drie of vier clusters.

Wat ziet Koopmans als zij ernaar kijkt? „Hiermee kun je meteen zien of de groep mensen die ziek is in een land allemaal hetzelfde virus hebben. En dat is hier in Nederland dus duidelijk niet het geval. Daarnaast zien we in de Nederlandse clusters ook een paar sequenties zitten uit andere landen. Dat refereert eraan dat dezelfde clusters ook bestaan in bijvoorbeeld Italië en Frankrijk. Dat is eigenlijk goed nieuws, want het betekent dat de Nederlandse virussen nog steeds getraceerd kunnen worden als introducties uit andere landen. We hebben hier nu de tweede, derde generatie van coronavirussen in Nederland en omdat ze nog niet heel ver weg zijn van de oorspronkelijke virussen denk ik dat de maatregelen die het RIVM en de overheid nu nemen nog effect kunnen hebben op de epidemie.”
Animatiefilm
Maar het is wel iets om goed in de gaten te houden, benadrukt Koopmans. „Naarmate het virus zich dag op dag verspreidt in één regio krijg je een steeds sterker regiosignaal, een cluster van virussen die sterk op elkaar lijken. Dan zijn ze niet langer sterk gelinkt aan virussen uit brongebieden. Wij blijven daarom sterk letten op of er Netherlands only-clusters ontstaan. Als dat gebeurt, is de epidemie hier in een andere fase gekomen.”
De groei van de stamboom en de verspreiding van het virus laten zich op de website van Nextstrain afspelen als een animatiefilm. Aan de hand van de genetica is het spoor van het virus te volgen. Dat is belangrijke informatie voor epidemiologen. Het geeft antwoord op vragen als: waar komen nieuwe besmettingen precies vandaan, en hoe lang circuleert het virus al in een bepaald gebied? Het rna van het virus fungeert als een moleculaire klok, die ‘tikt’ gemiddeld met een snelheid van iets meer dan één mutatie per maand. Die kleine verschillen en het patroon van de veranderingen geven genetici informatie over waar en wanneer bepaalde varianten kunnen zijn ontstaan.
De software van Nextstrain, ontwikkeld door de Amerikaanse bioinformatici Trevor Bedford en Richard Neher, brengt dat ook voor niet deskundige ogen goed in beeld. Op de website van Nextstrain zie je Sars-CoV-2 in drie maanden tijd vanuit China telkens in nieuwe varianten afsplitsen, aangegeven als een boom met veelkleurige takken.
De verspreiding van dit nieuwe virus is ongelooflijk hard gegaan. Omdat het virus helemaal nieuw is, heeft geen mens afweerstoffen ertegen. En er is ook nog geen vaccin dat het kan stoppen. Isolatie is de enige doeltreffende methode om het virus tegen te houden. Ondanks ingrijpende maatregelen in China en andere landen die dit probeerden te bewerkstelligen, lukte het niet het virus te stoppen. Geen land zal meer aan het virus ontsnappen.
Hoe nauwkeurig Nederland het virus kan volgen via deze genetische vingerafdrukken, is ook afhankelijk van hoe goed buitenlandse collega’s hun gegevens delen. Dat kan nog wel een stuk beter. In de databank van Gisaid waaruit Nextstrain zijn gegevens put, zijn tot nu toe meer dan 400 sequenties van SARS-CoV-2 opgenomen. Is dat niet erg weinig, aangezien het aantal bevestigde besmettingen wereldwijd is opgelopen naar meer dan 130.000? „Ja dat is heel weinig op het totaal”, zegt Emma Hodcroft van Nextstrain aan de telefoon vanuit Zwitserland. „We doen het sequencen nooit zelf”, zegt Hodcroft, die werkt als developer bij Nextstrain. „We zijn afhankelijk van andere onderzoekers die hun resultaten delen. Hoe meer sequenties we hebben, hoe meer we bij elkaar kunnen puzzelen. Maar er zijn nog witte plekken op de kaart, ook op plaatsen waar nu grote uitbraken aan de gang zijn, zoals Iran. Dat maakt het lastiger om connecties te zien tussen nieuwe besmettingshaarden.”
Ook het beeld van de Europese epidemie is gebrekkig. „We hebben lang niet zo veel samples als we zouden willen uit Europese landen”, zegt Hodcroft, „maar we blijven hoopvol dat die nog zullen komen. We weten vaak wel dat landen de sequenties al verzameld hebben, maar het gaat er natuurlijk om dat ze die ook met ons delen.”
Op basis van weinig datapunten is het lastig „één verhaal” te maken. Zo bevat de databank één virusgenoom gedateerd op 28 januari van de kleine, vroege uitbraak bij een auto-onderdelenbedrijf in Beieren en twee sequenties van virussen uit het begin van de uitbraak in Lombardije in de tweede helft van februari. Deze Duitse en Italiaanse virussen leken als twee druppels water op elkaar. Daarop suggereerde Nextstrain-oprichter Trevor Bedford in een tweet dat een niet-ingedamd virus uit de vroege Beierse uitbraak mogelijk de kiem was geweest voor de rampzalige Italiaanse uitbraak. Dat kwam hem onmiddellijk te staan op een storm van verontwaardiging van collega’s.
„Ja dat was een onfortuinlijke misstap”, zegt Hodcroft. „Hij had in zijn tweet niet goed duidelijk gemaakt dat er natuurlijk twee scenario’s zijn die elk op zich waar kunnen zijn. Dat de virussen zo op elkaar lijken kan betekenen dat zij in dezelfde besmettingsketen zitten, waarbij het virus dus van Beieren naar Italië is gegaan. Maar het andere scenario is dat de besmettingen in Duitsland en in Italië toevallig beide zijn begonnen met een overdracht van het virus uit China.”
Hoewel overinterpretatie dus snel op de loer ligt, kan het onderzoek van Nextstrain ook wilde theorieën snel ontkrachten. Dat gebeurde bijvoorbeeld toen Chinese onderzoekers stelden dat het virus zich aan het opsplitsen was in twee afzonderlijke stammen. De ene stam zou een stuk agressiever en dodelijker zijn dan de andere. Op basis van hun databank konden de wetenschappers van Nextstrain daar op hun beurt al gauw gehakt van maken.
Amerikaanse epidemie
De genoomanalyse bewees ook zijn kracht in de Verenigde Staten, waar heel lang werd gewacht met het testen van mensen op het nieuwe coronavirus. Trevor Bedford constateerde dat het virus in zijn eigen omgeving rond Seattle al heel lang ongemerkt moest rondwaren, nadat hij ontdekte dat een virusgenoom van 27 februari wel heel erg leek op de sequentie van een virus dat al zes weken eerder in Washington State was aangetroffen bij een man die was teruggekeerd vanuit Wuhan. Inmiddels hebben testen en andere virussequenties verzameld bij patiënten bevestigd dat er in de noordwestelijke staat een omvangrijke epidemie gaande is, waarbij al dertig doden zijn gevallen.
Een andere opvallende conclusie die je uit de gegevens in de Nextstrain-software kunt halen, is dat de virusvarianten waar de Verenigde Staten nu mee kampen, hoofdzakelijk níét uit Europa komen. De maatregel van de Amerikaanse president Trump om alle luchtreizigers uit de Schengenlanden te weren is gebaseerd op de verkeerde aanname dat de Amerikaanse besmettingen uit Europese bron kwamen. Nu zal de maatregel Europa in de toekomst wellicht beschermen tegen overdracht uit de VS, als de epidemie daar erger wordt.
Dankzij de genoomdatabank van het nieuwe coronavirus is ook een goed beeld ontstaan van wanneer het virus naar de mens oversprong. Aan de hand van de moleculaire klok kon dat moment teruggerekend worden naar medio november 2019. Dat klopt ongeveer met de eerste melding van een patiënt met een „onbegrepen longziekte” op 1 december in de Chinese stad Wuhan, het epicentrum van waaruit de epidemie zich verspreidde. „De vroegste virusvolgorde in onze bestanden is van 21 december 2019, verkregen uit een monster van een 65-jarige man in Wuhan die besmet bleek met het nieuwe virus. Op die datum was nog helemaal niet bekend om wat voor virus het ging, laat staan dat het virus al geïsoleerd was. Maar de sequentie is later alsnog toegevoegd toen het virus in een bewaard gebleven monster gesequencet werd.”
De geschiedenis is interessant, maar de focus is nu vooral op de actuele situatie, zegt Hodcroft. „We willen het liefst sequenties van recente monsters erbij zodat we de epidemie zo dicht mogelijk bij het moment van nu kunnen volgen.”
Het sequencen van een virusgenoom is specialistisch werk en kost ook tijd, waardoor je analyse achterloopt op de actualiteit. Niet bij iedereen die positief is getest kan ook een genoomvolgorde van het virus bepaald worden, zegt Koopmans. „Dat hangt af van de hoeveelheid virus die iemand bij zich draagt.” Uiteraard zou ze ook graag meer sequenties uit andere Europese landen erbij hebben, om de Nederlandse uitbraak beter in perspectief te kunnen zien. Immers, bij elk nieuw cluster dat aan de databank wordt toegevoegd, verschuiven de takken in de boom. „Dat is precies waarom je erg voorzichtig moet zijn met het trekken van conclusies”, zegt Koopmans, „Maar: met meer sequenties wordt de analyse wel beter.”
Aanvulling 21:17: In een eerdere versie van dit artikel stelde viroloog Marion Koopmans dat de epidemie nog in te dammen was. Mede in het licht van het interview met RIVM-directeur Jaap van Dissel in NRC, waarin hij stelt dat de uitbraak in Brabant niet meer in te dammen is, zijn die passages hierboven aangepast.
Virus-stambomen
Het spel met de kralen
Het erfelijk materiaal van het nieuwe coronavirus bestaat uit rna. Dat rna ligt als een lang kralensnoer van 30.000 ‘letters’ in de omhullende mantel van het virusdeeltje. Het rna bevat alle instructies voor het maken van een nieuwe generatie virusdeeltjes, inclusief alle manteleiwitten. Het virus gebruikt de machinerie van de gastheer voor het maken van vele kopieën van zichzelf, die vervolgens weer vrijkomen en op hun beurt nieuwe cellen infecteren.
Maar het kopiëren van het virus gaat niet foutloos, omdat het virus niet kan controleren of de gemaakte kopie exact hetzelfde is als het origineel. Soms wordt er een verkeerde letter ingebouwd in het rna, of worden er zelfs hele stukken overgeslagen. Door dit slordige kopiëren krijgen virussen na verloop van tijd een unieke genetische vingerafdruk.
De software van Nextstrain berekent hoe de stamboom er moet uitzien op basis van de verschillen in de vingerafdrukken, rekening houdend met de mutatiesnelheid van het virus. Die is gebaseerd op het getal van andere coronavirussen, en tot nu toe zijn er geen aanwijzingen dat SARS-Cov-2 daarvan zou afwijken. Het komt erop neer dat iedere genetische letter in de code gemiddeld eens in de 2.000 jaar zal veranderen. Dat is een heel normale snelheid voor virussen, ook het ebolavirus en het zikavirus zitten hier in de buurt. Maar er zijn uitzonderingen; bijvoorbeeld het griepvirus verandert twee tot vier keer sneller.